Colab notebooks, Docker images, and Snakemake workflows I use for reproducible bioinformatics (PhosBoost, structure tools, RNA-seq processing, containers for docking and PPI exploration). Everything below used to live on separate pages; this is the combined reference.
Table of contents
Colab notebooks
I have been working on creating Colab notebooks for a number of bioinformatics projects that I have been working on.
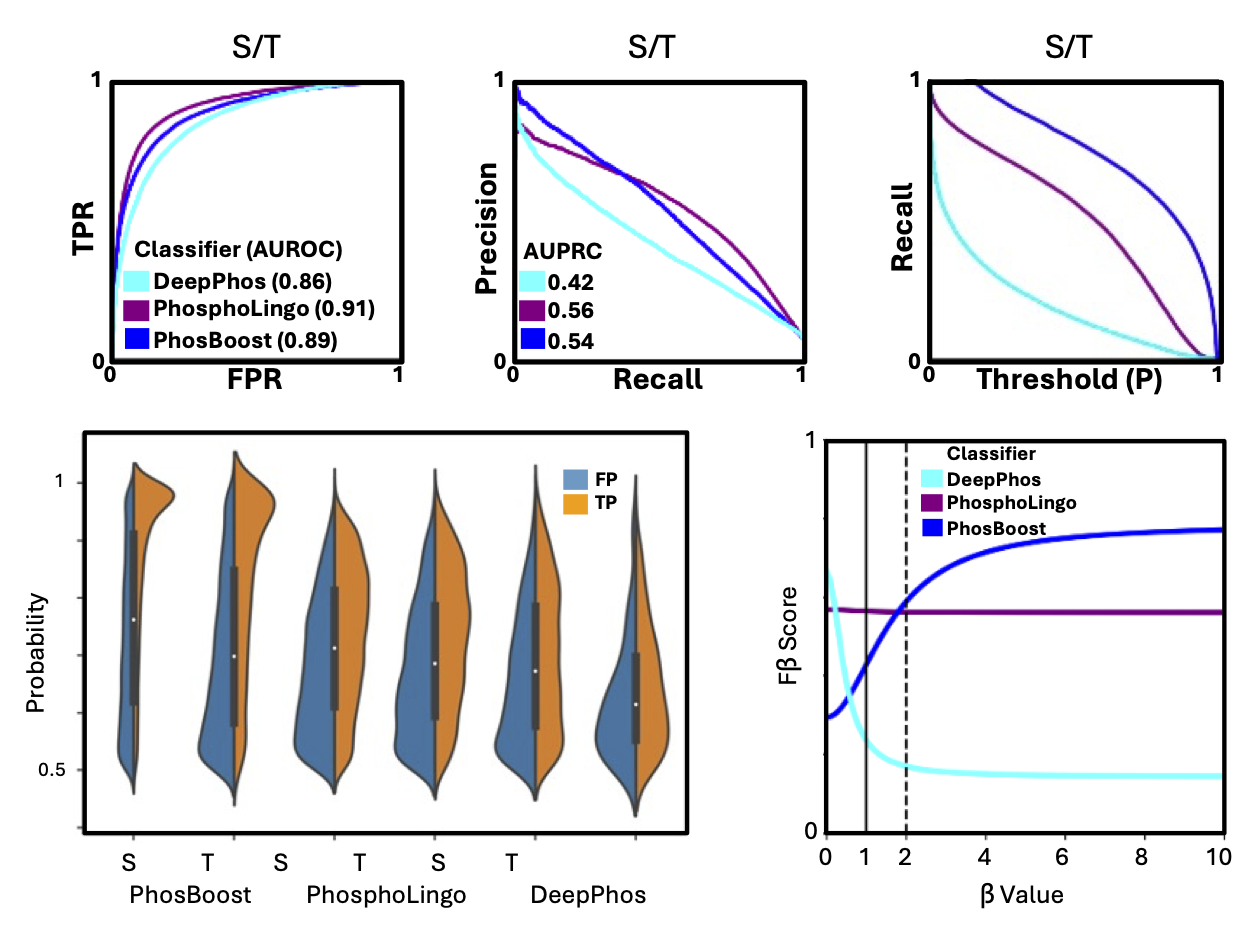
PhosBoost
![]()
A Colab notebook I prepared for predicting protein phosphorylation sites for a user-provided protein, based on our PhosBoost manuscript. The Colab notebook runs fast on a CPU node, so it should also work with a free account. Additional details can be found in the paper, GitHub repository, and the Colab notebook.
AllMetal3D
![]()
A minimal Colab notebook for predicting metal and water binding sites in proteins. Works with CUDA out of the box when running on a GPU node.
AlphaFill-Relax
![]()
A Colab notebook I prepared for running a brief 20ps Molecular Dynamics (MD) equilibration simulation step using OpenMM engine and AMBER force field. My very preliminary results show that it can improve the positioning of metal cofactors in terpene synthase active sites. Additional context is on the protein–ligand tutorial and inside the notebook.

Relax Amber Batch
![]()
A Colab notebook I prepared for running the amber relaxation step recommended for predicted protein structures. This notebook is based completely on the relax_amber notebook that can be found at the ColabFold repository. The only addition offered here is the ability to batch-apply the PDB relaxation step to all PDB files in a given Google Drive folder.

Docker images
I have been working on creating Docker images for a number of bioinformatics tools that I used on a regular bases. In addition to just having functional Docker images to run different workflows, I am also adding step-by-step instructions on how to access the Docker container and how to generate the input and output files using each tool. Below, you can find instructions on how you can make your own Docker images and brief description of the Docker images I created.
DiffDock
DiffDock can be used to run molecular docking analyses of specified small molecules and a given protein structure.
AlphaFill
AlphaFill can be used to transplant ligands from experimental structures to predicted structures based on structure similarity. This AlphaFill Docker image can be used to run the stand-alone version of AlphaFill. Additional instructions and details are provided in the Docker Hub page.
P2Rank
PanPPI
| A Python Dash web-app that I built to explore the PanPPI data we generated using the maize pan-genomes, as explained in this [G3: Genes | Genomes | Genetics](https://academic.oup.com/g3journal/advance-article/doi/10.1093/g3journal/jkae059/7630293) paper. Currently there is only a Docker image for the Dash app with the maize data included, but I am planning to add a Docker image for running the analysis workflow for custom pan-genomes and custom data. |
PhylogenyZ
An environment to run simple phylogenetic analyses, meant primarily for building protein trees. The general workflow I use is: (1) protein alignment using FAMSA, (2) alignment trimming using ClipKIT, and (3) building the phylogenetic tree using IQ-TREE2.
Chainsaw
An Docker image for running Chainsaw, a protein domain segmentation based on a given PDB structure.
Evolocity
An Docker image for running Evolocity, a Python package that calculates evolutionary velocity using protein languages. This Docker container comes with the default esm1b_t33_650M_UR50S and esm1b_t33_650M_UR50S-contact-regression pre-trained models downloaded so it takes a bit more space.
Snakemake
The latest snakemake files can be found in my github bioinformatics-repo repository in the GitHub repo. Additional information for specific workflows is provided in the specific sections below.
Snakemake — RNA-seq
The goal is to create a couple of snakemake workflow files that take care of obtaining and processing RNA-seq data. Each file contains.
For processing RNA-seq data it makes more sense to run the workflow one sample at a time. In snakemake it is possible to tranverse DAG depth-first as explained here. It is not necessary to run workflow this way, but that is how I am writing all my RNA-seq processing workflows.
1. Set a priority for each rule starting with 1 for the first rule
2. Call snakemake with the following argument: -j 1
Requirements
I use a clean conda environment to run snakemake workflows. I ran into errors in the past that were due to conda not installing the latest repositories of some packages by default. That is why I specify which version to install now. I still have trouble installing the latest available packages in some cases. I will include a conda spec-file with the exact environment that I use but until then I am including some the steps that are needed to run the workflows.
conda install -c conda-forge genozip
conda install -c conda-forge libgcc-ng=11.2.0
conda install -c bioconda fastp=0.23.2
conda install -c bioconda hisat2=2.2.1
conda install -c bioconda sambamba=0.8.2
conda install -c bioconda subread
Finally, I write all of the intermediate files on a ramdisk before writing them on the hard-drive. It does require having a large amount of RAM (>96gb) but it significantly reduces the wear and tear on the SSD drive and reduces analysis time by avoiding unnecessary compression/decompression steps. The 2 lines of code below require linux sudo privilege and will mount a ramdisk folder. That said, a mounted ramdisk is not required, only a folder named “ramdisk” is required.
# To create a ramdisk folder
mkdir ramdisk
sudo mount -t tmpfs -o size=95000m tmpfs ramdisk/
Getting the metadata for an RNA-seq BioProject
I prefer working with the European Nucleotide Archive (ENA - https://www.ebi.ac.uk/ena/browser/home) for all analysis steps. If you want to process a published RNA-seq dataset, a good place to start is by downloading the metadata for a BioProject using the Study Accession (PRJNAXXXXXX). I have made a python script that downloads the metadata using the ENA API, based on their data accessing tutorial. The script takes the Study Accession as an argument and saves the metadata as a CSV file with the ID name. The CSV file should contain all the needed information, inclduing sample descriptions and FTP download links.
# Download BioProject metadata from ENA
python ena_metadata_download.py PRJNAXXXXXX
From an SRA/ENA BioProject ID to count files
This snakemake workflow does not require an existing ENA metadata file, just a Study Accession, a HISAT2 genome index and a genome GTF file. The index and GTF files should have the same name
snakemake -F -p -j1 --keep-going --snakefile all2counts_ena.smk --config sra=PRJNA123456 idx=Zmays_493_APGv4_Phytozome