During my Ph.D. in the Huffaker lab and a Post-doc in the Schmelz lab at UC San Diego, I conducted research on different aspects of plant immune responses against pests and disease. My work focused on understanding the maize perception of endogenous peptide hormones and herbivore-associated molecular patterns, and studying a class of antibiotic specialized root sesquiterpenoid metabolites and their role in mediating interaction with root microbes. I have also contributed on a number of research projects, providing extensive bioinformatics analysis results and conducting experiments. As part of my bioinformatics work, I have analyzed RNA-seq data, constructed phylogenetic trees, conducted QTL and GWAS mapping, used variant calling to generate an improved SNP-based genetic map, and generated comparative genomic analyses of syntenic gene clusters. I have also built a Shiny web-application for Mutual Rank-based gene coexpression analysis, named MutRank, that was instrumental in elucidating the biosynthetic pathways of maize specialized metabolism.
Table of contents
- Differential activities of maize plant elicitor peptides as mediators of immune signaling and herbivore resistance
- Comparative analyses of responses to antiherbivore elicitors to identify maize gene candidates mediating sensitivity to FACs
- MutRank, an R shiny web-app for exploratory targeted mutual rank-based coexpression analyses
Differential activities of maize plant elicitor peptides as mediators of immune signaling and herbivore resistance
One of the first bioinformatics projects I have ever worked on was the analysis of microarray gene expression data from maize samples treated with 11 different combinations of maize plant elicitor peptides (Peps). Peps are conserved regulators of defense responses and models for the study of damage-associated molecular pattern-induced immunity, found across almost all plant families. Although present as multigene families in most species, with a fairly large peptide sequence diversity, the functional relevance of these multigene families was largely undefined. To better define the function of individual maize Peps and their cognate receptors (ZmPEPRs), we used different approaches to measure their activities, including changes in defense-associated phytohormones, specialized metabolites and global gene expression patterns, in combination with heterologous expression assays and analyses of CRISPR/Cas9-generated knockout plants. Our results showed that the different Peps behave similarly, and that their specific activities are defined by the relative magnitude of their response output rather than by uniqueness. Another discovery was that the maize Pep precursors contain multiple active Peps, a phenomenon not previously observed for this family.

Another discovery that we made, was that the inactive ZmPep5a might actually function as an antagonist to Pep-induced responses, a regulatory mechanism not previously observed for this family. Using ligand–receptor modeling of the bioactive ZmPep3 and inactive ZmPep5a revealed structural features critical to the function of ZmPeps and showed that both Peps are predicted to bind to the same region on the maize Pep-receptor. The biological relevance of the ZmPep5a antagonist activity is yet to be determined.

Comparative analyses of responses to antiherbivore elicitors to identify maize gene candidates mediating sensitivity to FACs
Maize responds to herbivore-associated molecular patterns (HAMPs) found in the oral secretions of attacking caterpillars by activating dynamic direct and indirect antiherbivore defense responses. For example, the elicited production of volatile organic compounds (VOCs) emitted from leaves following caterpillar herbivory can attract parasitoid wasps and enhance parasitization rates, as an indirect defense response. In the photo below, you can see the set-up for collecting volatiles after an overnight treatment with anti-herbivory defense elicitors.

Maize has been a model for HAMP identification, using volatile bioassays to measure HAMP activity, leading to the discovery of the first defined biomolecules from insect oral secretions, termed fatty acid–amino acid (FACs). FACs are a family of molecules based on the conjugation of plant-derived linolenic acid to either glutamine or glutamate in the insect gut. My work focused on the N-linolenoyl l-glutamine (Gln-18:3), one of the most highly abundant and potent FAC elicitors, while using an anti-herbivory peptide hormone, ZmPep3, as an experimental control. While we knew that FACs and Peps activate similar antiherbivory defense responses in maize, the RNA-seq experiment I conducted provided really strong evidence that they function through a similar signaling pathways. Unfortunately, because they both differentially regulate quite a lot of genes, the RNA-seq experiment didn’t help us with identifying candidate genes involved in FAC-induced responses.

After finding two maize inbreds with differential sensitivity to FACs, but not to ZmPep3, we decided to use a forward genetic mapping approach. We used the maize IBM bi-parental mapping population to conduct a QTL experiment based on the differential response of the two maize inbred parents, B73 and Mo17, which led to the identification of a single locus on chromosome 4 associated with FAC-sensitivity.
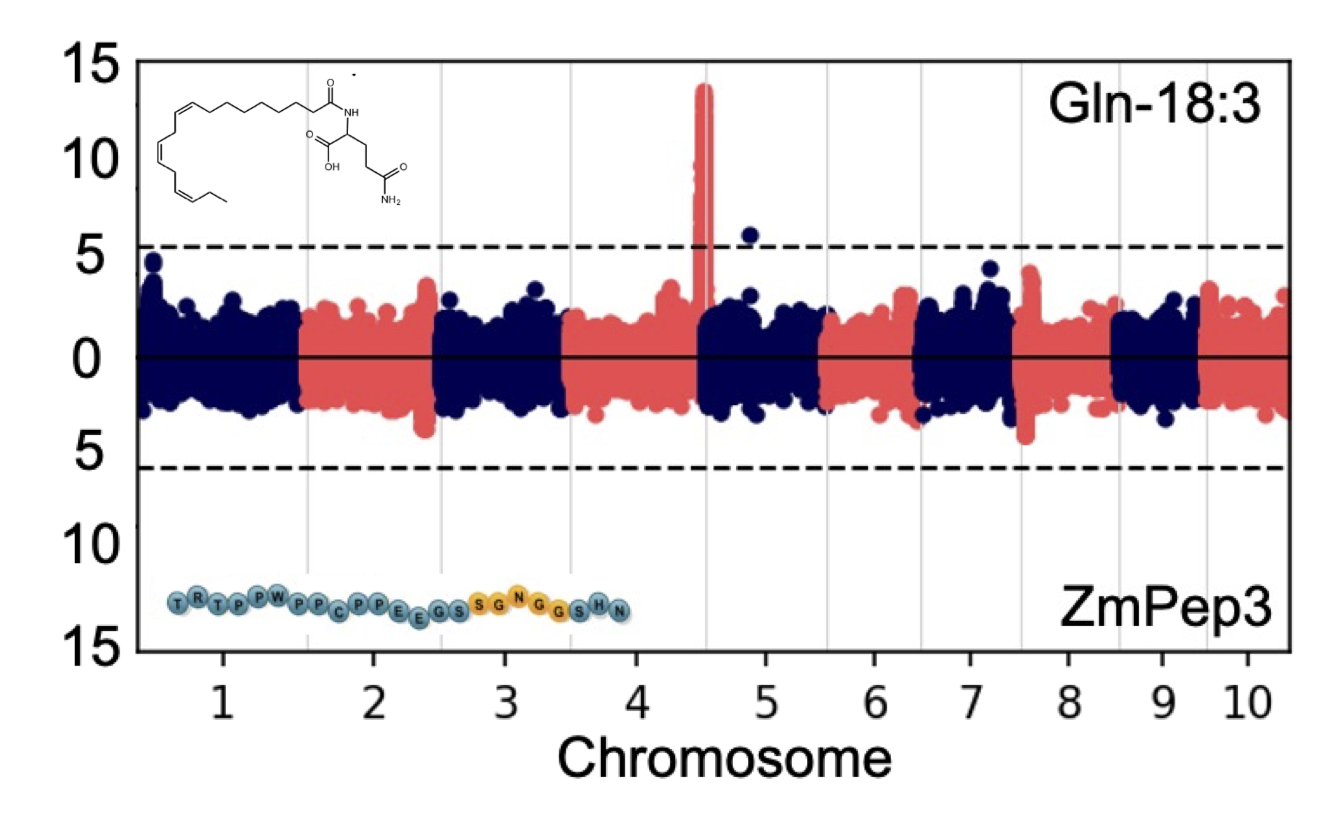
Additional fine-mapping approaches, including using a new RNA-seq-based SNP map I generated, helped us to further narrow the locus to 19 candidate genes. The top candidate gene identified, termed FAC SENSITIVITY ASSOCIATED (ZmFACS), encodes a leucine-rich repeat receptor-like kinase (LRR-RLK) that belongs to the same family as a rice (Oryza sativa) receptor gene that was previously found to be associated with herbivory resistance. Consistent with reduced sensitivity, qPCR analysis of ZmFACS expression in B73 and Mo17 showed that the gene had significantly lower expression in Mo17 compared to B73. Transient heterologous expression of ZmFACS in Nicotiana benthamiana resulted in a significantly increased FAC-elicited response, suggesting that ZmFACS is involved in FAC-induced responses in maize.

MutRank, an R shiny web-app for exploratory targeted mutual rank-based coexpression analyses
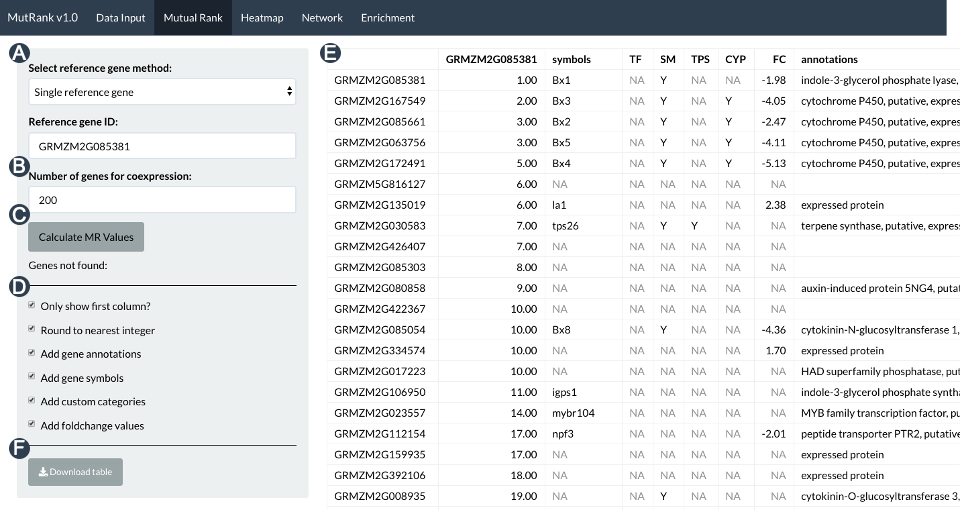
This work builds upon the work in a paper by Jennifer Wisecaver that showed that gene coexpression analysis using mutual rank is highly effective at detecting coregulated genes across different plant specialized metabolism pathways. Mutual rank, first described in a paper by Takeshi Obayashi, is calculated as the geometric mean of the rank of the Pearson’s correlation coefficient between a pair of genes. The R Shiny MutRank app was built with the specific goal of making mutual rank-based gene coexpression analyses simple, customizable and user-friendly to enable the analysis for researchers with minimal bioinformatics experience. This app has proven to be useful and contributed extensively to the elucidation of different plant specialized metabolism pathways, including the maize Kauralexin and Zealexin pathways.