Table of contents
PhosBoost
![]()
PhosBoost is a machine learning approach that leverages protein language models and gradient boosting trees to predict protein phosphorylation from experimentally derived data. PhosBoost offers improved performance when recall is prioritized while consistently providing more confident probability scores.
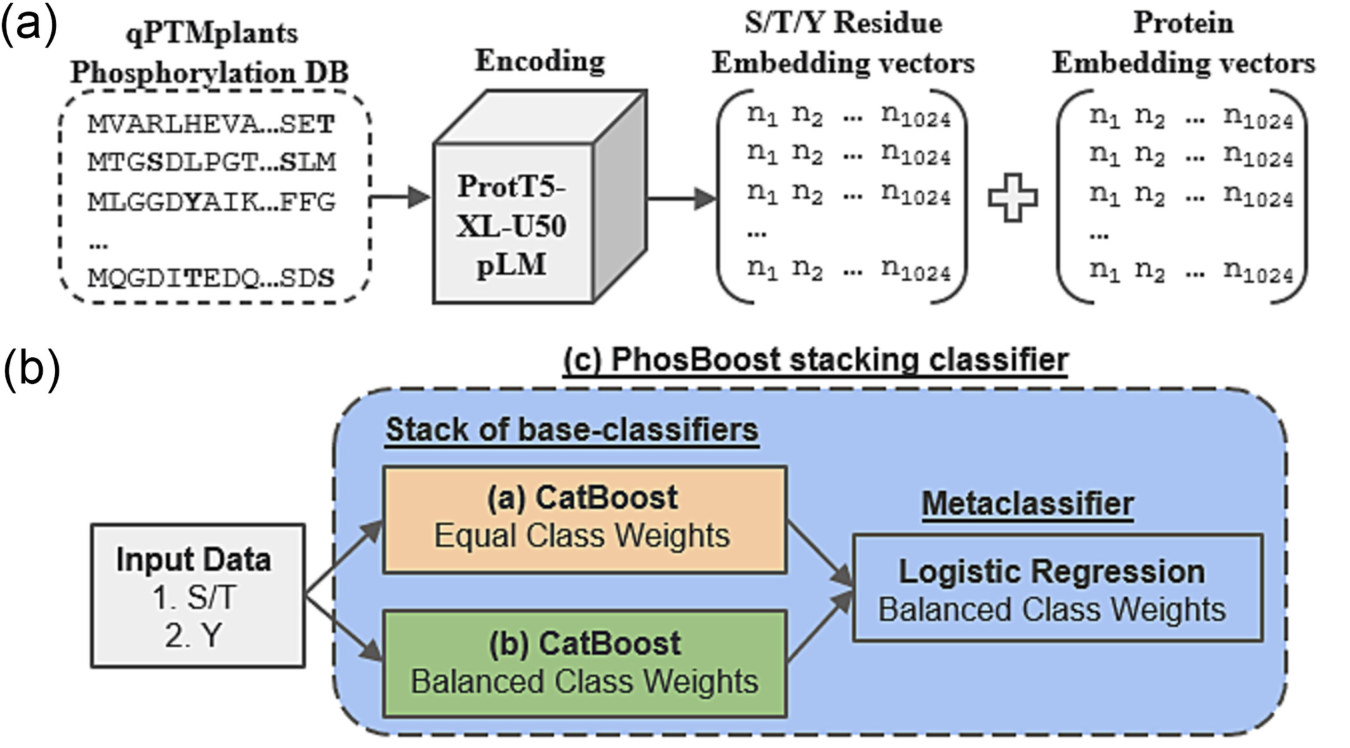
Poretsky, E., Andorf, C. M., & Sen, T. Z. (2023). PhosBoost: Improved phosphorylation prediction recall using gradient boosting and protein language models. Plant Direct, 7(12), e554.
PanPPI
In the PanPPI framework, we generated predicted STRING-db interactomes for the 26 maize NAM inbred lines and used ClusterONE to cluster the genome-, core-, and pan-interactomes. The clusters were then annotated using GO term enrichment, gene coexpression, and gene descriptions. The annotated clusters can be used putative gene function predictions and prioritization of candidate genes. The framework can be applied to any list of pan-genomes, see the GitHub repository for instructions. We also generated a Python Dash web-application to help with finding relevant PPI clusters with user-provided genes of interest. The easiest way to access the app, with the maize data pre-loaded, is by following the instructions in the provided Docker page (instructions to install Docker). Alternatively, the code and instructions for the Dash app are available in the GitHub repository.
G3: Genes, Genomes, Genetics GitHub Docker
Poretsky, E.*, Cagirici, H. B.*, Andorf, C. M., & Sen, T. Z. (2024). Harnessing the predicted maize pan-interactome for putative gene function prediction and prioritization of candidate genes for important traits. G3: Genes, Genomes, Genetics, jkae059.
Structural Variability of Pfam Domains
This work combines AlphaFold2-predicted structures with Pfam domain annotations to characterize structural variability within domain families. The PfamFold workflow extracts domain coordinates, computes structural features, and uses FoldSeek together with agglomerative clustering to group similar domain conformations—useful for interpreting domain diversity and for curation of Pfam families.
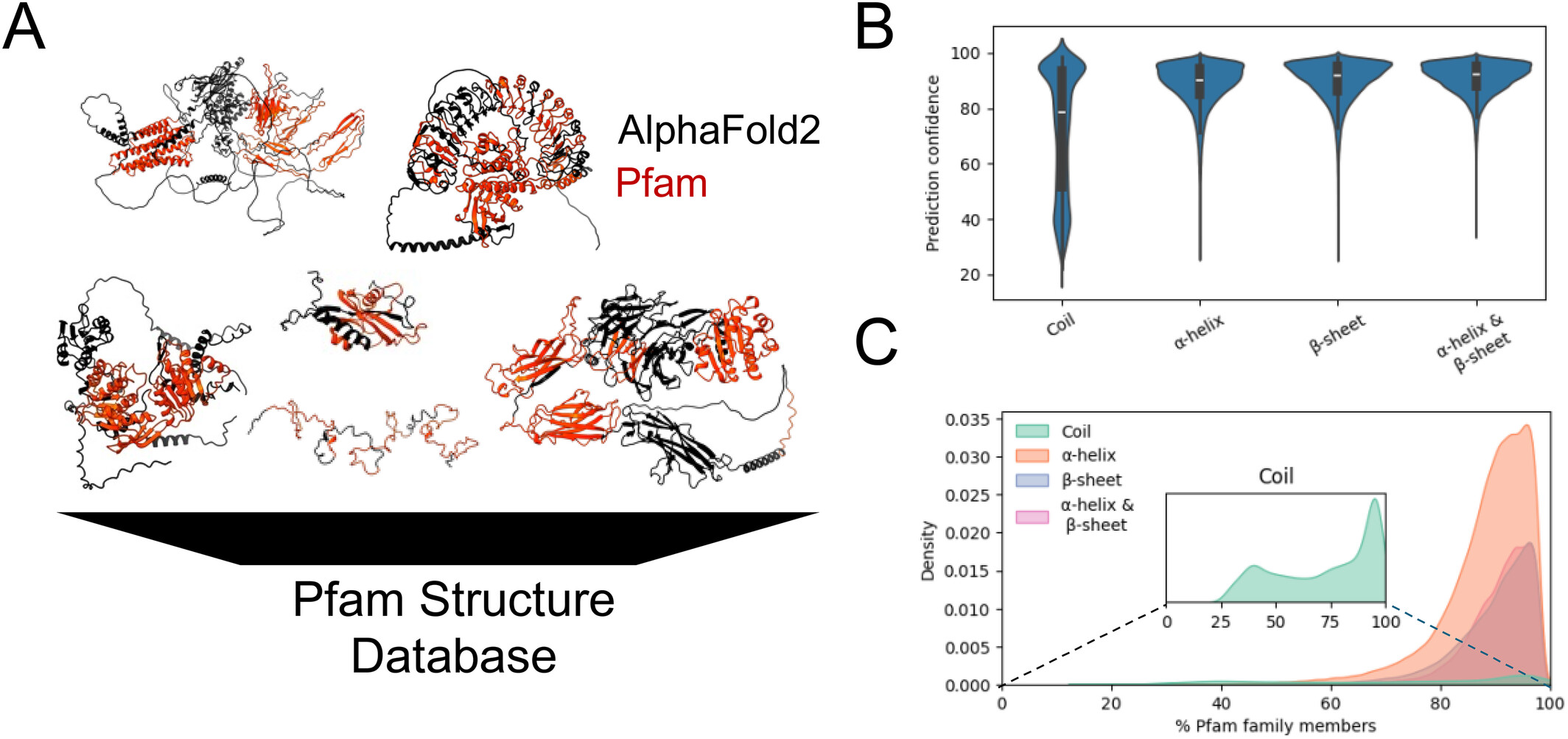
Poretsky, E., Andorf, C. M., & Sen, T. Z. (2025). Structural variability of Pfam domains based on Alphafold2 predictions. Proteins, 93(12), 2182–2192.
LLMs in Biocuration
Large language models (LLMs) can help extract structured information from the scientific literature, but their accuracy must be measured against expert curation. In the ChatMTA project we compared GPT-3.5 and GPT-4 to a professional curator on wheat and barley genetic mapping papers, using a retrieval-augmented QA setup to probe traits and QTLs. The study highlights where LLMs are already useful for biocuration workflows—and where human review remains essential.
Poretsky, E.*, Blake, V. C., Andorf, C. M., & Sen, T. Z. (2025). Assessing the performance of generative artificial intelligence in retrieving information against manually curated genetic and genomic data. Database, 2025, baaf011.
Ligand Docking
LiganDock is a Python toolkit for automated receptor and ligand preparation, flexible grid-box definition (marker ligand, selected residues, or PyKVFinder pockets), and batch docking with AutoDock Vina—wired to Meeko, RDKit, OpenBabel, and related tools. I am still interested in applying docking and related structure-based methods (including approaches such as DiffDock and DynamicBind) to predicted plant specialized metabolism enzymes, especially terpene synthases. Related notes and resources also appear on the protein-ligand page.