This section includes code for processing and analyzing different aspects of protein structures.
Table of contents
AlphaFill
Transplanting ligands
We are going to use the ColabFold predicted structure of the grape UGT72B27 uridine diphosphate-dependent glycosyltransferase (UGT) as an example. The AlphaFill webserver can be used to transplant a uridine diphosphate (UGT) ligand from the experimental 2VG8 structure. The code below can be used to convert the AlphaFill CIF file to a PDB file. We want to generate a PDB file because many tools for analyzing protein-ligand interactions, such as PLIP, work better with PDB-formatted files. We will use OpenBabel to convert the CIF file to PDB, and then use ProDy to edit the PDB file so it is compatible for PLIP-based analysis, but depending on which tools you intend to use, the PDB file might need to be modified in different ways. Download the ColabFold predicted structure of UGT72B27 here. Submit the PDB file to the AlphaFill webserver, select the UGT ligand only, and download the AlphaFill CIF file as shown in the image below.

from openbabel import openbabel
import prody as pr
# Convert the AlphaFill CIF file to a temporary PDB file using OpenBabel
prot_id = "UGT72B27"
obConversion = openbabel.OBConversion()
obConversion.SetInAndOutFormats("cif", "pdb")
mol = openbabel.OBMol()
obConversion.ReadFile(mol, '{}.afill.cif'.format(prot_id))
obConversion.WriteFile(mol, '{}.afill.tmp.pdb'.format(prot_id))
# Load PDB file using ProDy
structure = pr.parsePDB('{}.afill.tmp.pdb'.format(prot_id))
# Get the ResNames of all ligands in the PDB file
# In this example, the res_name_list only contains a single ligand: 'UGT'
res_name_list = list(set(structure.select("not protein").getResnames()))
print(res_name_list, len(res_name_list))
# Find the last protein Resnum so we can set a larger ligand Resnum
max_res = structure.select('protein').getResindices().max()
# In this example we assume that there is only one ligand (UGT) in the PDB file
# We can use ProDy to iterate over every ligand atom and make the appropriate edits
res_name = "UGT"
for chain in structure.select('resname {}'.format(res_name)):
# Change the flag from ATOM to HETATM for the ligand
chain.setFlag("hetatm", 'hetatm')
# Set the ligand chain ID to 'B', keeping the protein chain ID as 'A'
chain.setChid("B")
# Set the ligand Resnum to be max_res + 1
chain.setResnum(max_res + 1)
# Save the modified structure as the final PDB file
pr.writePDB('{}.afill.pdb'.format(af2_id), structure)
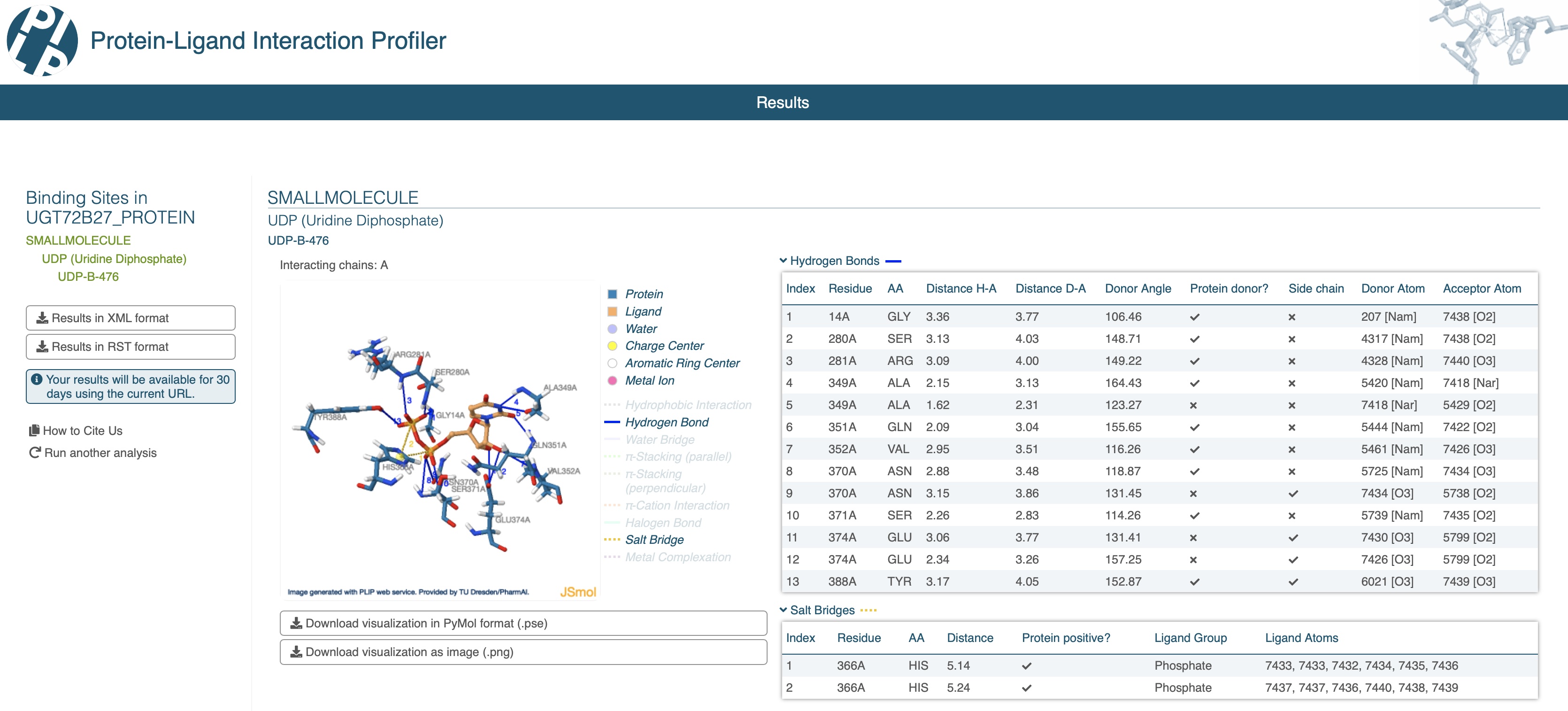
Now you can submit the modified AlphaFill PDB file (UGT72B27.afill.pdb) to the PLIP webserver to identify which protein residues interact with the different ligand atoms. As you can see in the image below, PLIP identified 13 hydrogen bonds and 2 salt bridges between the protein residues and the UDP ligand.
NOTE: AlphaFill does not guarantee that the transplanted ligand occupies the correct position and pose in the predicted structure, nor does it guarantee that it is an actual ligand for the predicted structure. The resulting AlphaFill structure and predicted interactions by PLIP should be carefully assessed.
Transplanting cofactors
In this case, we are going to use the AlphaFold2 predicted structure for the terpene synthase (TPS) O81192. The AlphaFill webserver can be used to transplant MG and MN metal cofactors from the experimental PDB structures, 1N1Z in this case. Because the AlphaFill database already analyzed the AF2 structures, we can just search for the O81192 AF2 ID and download the AlphaFill CIF structure with the MG and MN cofactors (skipping POP in this case). The code below can be used to convert the AlphaFill CIF file to a PDB file that is compatible with PLIP for analyzing protein-ligand interactions, and probably many other tools. We will use OpenBabel to convert the CIF file to PDB, and then use ProDy to edit the PDB file so it is compatible for PLIP-based analysis, but depending on which tools you intend to use, the PDB file might need to be modified in different ways. Submit the AF2 ID to the AlphaFill webserver, select the MG metal cofactors only (some TPS structures will have MN as well), and download the AlphaFill CIF file as shown in the image below.

from openbabel import openbabel
import prody as pr
# Convert the AlphaFill CIF file to a temporary PDB file using OpenBabel
prot_id = "O81192"
obConversion = openbabel.OBConversion()
obConversion.SetInAndOutFormats("cif", "pdb")
mol = openbabel.OBMol()
obConversion.ReadFile(mol, '{}.afill.cif'.format(prot_id))
obConversion.WriteFile(mol, '{}.afill.tmp.pdb'.format(prot_id))
# Load PDB file using ProDy
structure = pr.parsePDB('{}.afill.tmp.pdb'.format(prot_id))
# Select protein and MG/MN atoms from the temp PDB structure
structure = structure.select('protein or resname MG or resname MN')
# 1. Change the flag from ATOM to HETATM for MG/MN
# 2. Change Chain ID to A for all co-factors
# 3. Change the atom numbers to be above the last protein atom number
max_res = structure.select('protein').getResindices().max() + 1
for chain in structure.select('resname MG or resname MN'):
chain.setFlag("hetatm", 'hetatm')
chain.setChid("A")
chain.setElement(chain.getResname())
max_res += 1
chain.setResnum(max_res)
# Save the modified structure as the final PDB file
pr.writePDB('{}.afill.pdb'.format(af2_id), structure)
Now you can submit the modified AlphaFill PDB file (O81192.afill.pdb) to the PLIP webserver to identify which protein residues interact with the different ligand atoms. As you can see in the image below, PLIP identified a total of 4 interactions between the 3 MG metal cofactors and the protein residues. In the AlphaFill-Relax section below, I show an example on how you can run a short molecular dynamics equilibration step to try to get an improved model with more interactions between the MG metal cofactors and the protein residues.

NOTE: AlphaFill does not guarantee that the transplanted ligand occupies the correct position and pose in the predicted structure, nor does it guarantee that it is an actual ligand for the predicted structure. The resulting AlphaFill structure and predicted interactions by PLIP should be carefully assessed.
AlphaFill-Relax
![]()
AlphaFill transplants ligands from experimentally determined structures to predicted structures based on protein sequence and structure similarity. One of the limitations of AlphaFill is that it does not take into account the local environment into which the ligand is being transplanted. On the other hand, the predicted protein structure does not necessarily represent the optimal confirmation for ligand binding. While the transplanted ligands are placed with relatively good accuracy, it is likely that minor adjustments to the ligand positions and neighboring protein residues can further improve the accuracy of the model. With this assumption, it makes sense that adding a molecular dynamic (MD) simulation step could generate a more accurate representation of the protein-ligand interaction interface. Here is how a 20ps MD simulation of a terpene synthase with three Mg cofactor ligands looks -

I am calling this approach AlphaFill-Relax, to borrow from the AlphaFold-Relax method. The link above will take you to the Colab notebook, linked the jupyter notebook in my GitHub repository. As the explanation in the notebook says: This is a very preliminary approach that was not peer-reviewed nor extensively tested. It is completely based on the Making it rain: Cloud-based molecular simulations for everyone" (link to paper) Colab notebook for running Molecular Dynamics (MD) simulations using OpenMM engine and AMBER force field link to notebook, and uses just the Equilibration MD step with default parameters and a very short 20ps simulation time (taking a few minutes to complete using the T4 GPU node). Below is the before-and-after comparison of the Mg cofactor ligands that were transplanted to the AlphaFold2 structure of a terpene synthase O81086. From the animation above, it doesn’t look like the 20ps MD simulation has changed much, but an analysis of the original AlphaFill PDB file and the relaxed PDB file shows a large increase in the number of interactions (PLIP results, default parameters), including a better agreement with the conserved DDXXD motif involved in Mg binding.

As a note, I have only tested AlphaFill-Relax on metal cofactor ligands and have yet to test it on any other type of AlphaFill-transplanted ligands.