General research interests
Over the past decade, I have worked on diverse aspects of plant biology, genetics and bioinformatics analyses. Below is a sample of general research projects and questions that I often think about, with some relevant links. I describe my past research at UC San Diego and current research at the USDA in the Bay area in more detail in the pages linked in the sidebar.
Table of contents
- General research interests
- Developing ML models with protein language models (PLMs) and predicted structures
- Developing workflows for pan-genome protein-protein interaction networks
- Analysis of protein-ligand interactions using molecular docking and dynamic simulations
- Transcriptomic and gene coexpression analyses in plants
- Metabolomic profiling and multi-omic analysis of plant tissues
- Association mapping and the genetic basis for plant phenotypic diversity
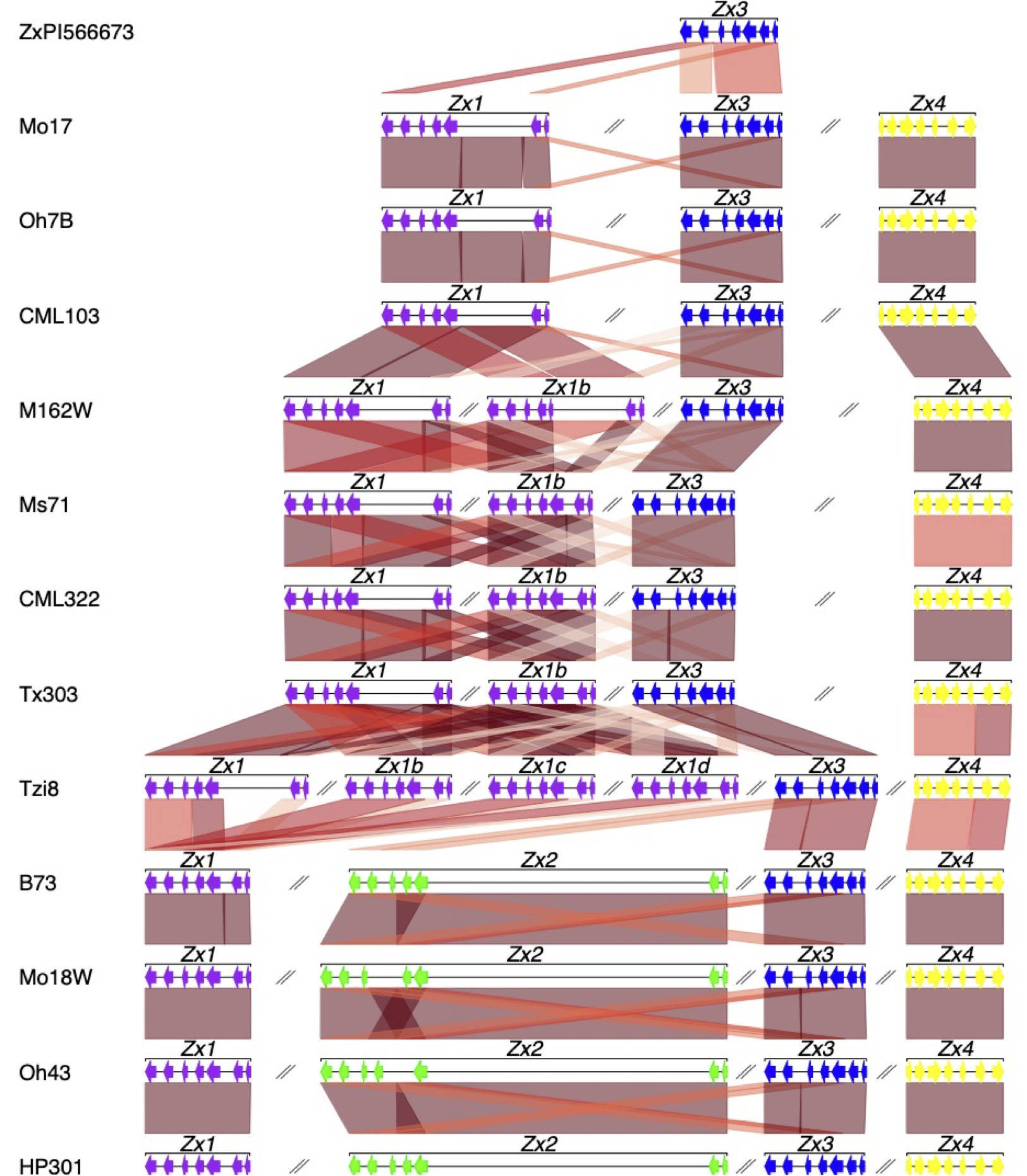
- Comparative genomic and gene synteny
Developing ML models with protein language models (PLMs) and predicted structures
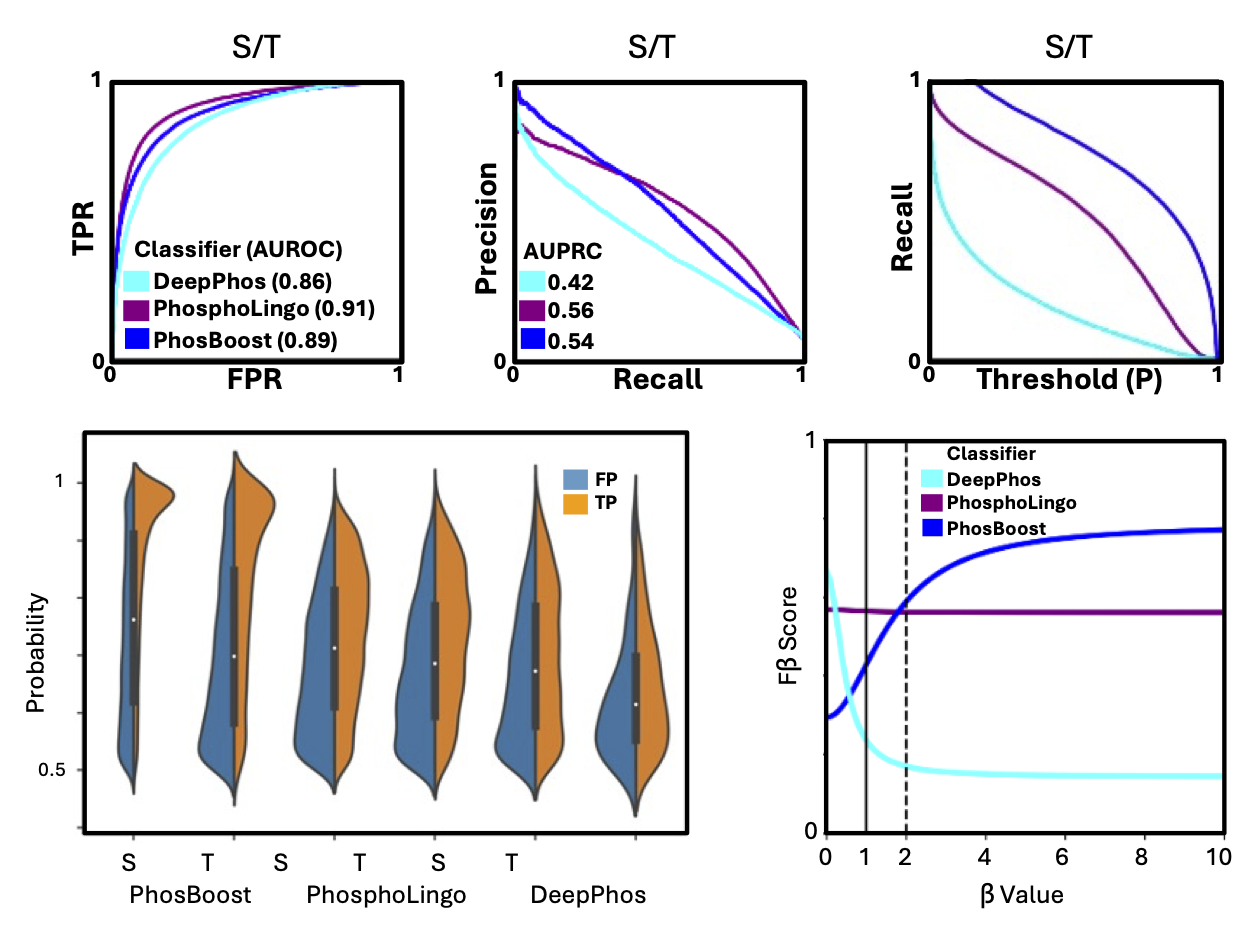
Protein phosphorylation is a post-translational modification that can alter the structure and function of proteins. PhosBoost is a machine learning approach that leverages protein language models and gradient boosting trees to predict protein phosphorylation from experimentally derived data. PhosBoost offers improved performance when recall is prioritized while consistently providing more confident probability scores. Additional details are available on the research/current page.
Developing workflows for pan-genome protein-protein interaction networks
We deployed the PPI clustering algorithm ClusterONE to identify numerous predicted-PPI clusters that were functionally annotated using gene ontology (GO) functional enrichment, demonstrating a diverse range of enriched GO terms across different clusters. We show that the functionally annotated PPI clusters establish a useful framework for protein function prediction and prioritization of candidate genes of interest. Additional details are available on the research/current page.

Analysis of protein-ligand interactions using molecular docking and dynamic simulations
Using predicted protein structures of metabolomic enzymes with generative molecular docking is a powerful tool for understanding substrate specificity and promescuity that enables the metabolomic diversity observed in plants. As part of my PhD and postdoc Past Research, I have contributed to multiple projects on maize specialized terpenoid antibiotic metabolies and led a project on understanding the biological role of root secreted sesquiterpenoid maize specialized metabolites. One of the interesting patterns that emerged from these research projects is the product promiscuity of some of the discovered terpene synthases (TPSs) and CYP450s. My current postdoc position at USDA, working on SOTA protein structure prediction and molecular docking methods, has sparked my interest in applying these methods to protein-ligand interactions in the context of plant specialized metabolism.

Transcriptomic and gene coexpression analyses in plants
Transcriptional reprogramming is one of the major initial rapid cellular changes that plant cells undergo in response to changing environmental cues and stress signals. For example, using ligand-induced differential gene expression (DGE), we have shown that maize responses to anti-herbivory peptide hormones and exogenous molecules from feeding caterpillar oral secretions induce highly overlapping transcription reprogramming responses, differing primarily in magnitude, rather than uniqueness. This observations suggest that maize immune responses against caterpillars are likely to be highly tuned and initiated through similar signaling pathways. Additional details can be found on the Past Research page. In addition to DGE analyses, I have have contributed to a number of collaborations studying maize specialized metabolism pathways using gene coexpression analyses. Specialized metabolism enzymes, including in plants, tend to be highly coregulated on the transcriptmic and proteomic level. We found that integrating gene coexpression analyses with forward genetic screens, such as QTL and GWAS mapping, can be used effectively to elucidate almost complete biosynthetic pathways. This part has lead to the development of MutRank, an R Shiny web-app, that greatly facilitates gene coexpression analyses. As part of the hypothesis generation process, I have processed hundreds of publicly available RNA-seq NGS samples, primaily from maize but not restricted to it, using an automated Snakemake workflow I developed to automate this process.

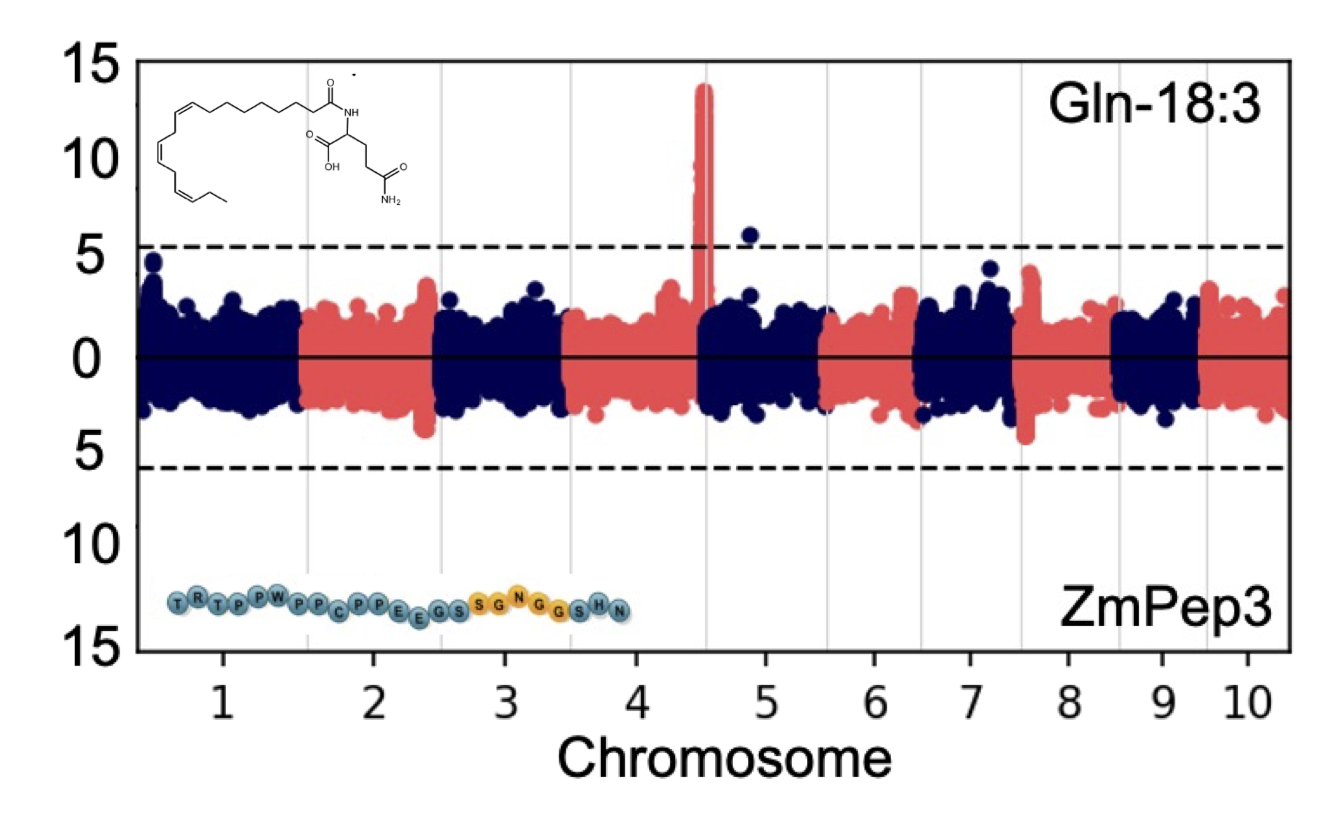
Metabolomic profiling and multi-omic analysis of plant tissues
Plant specialized metabolites play important roles in mediating beneficial interaction with microbes and insects and preventing damage from pests and disease. Extraction and profiling of plant metabolites is vital for untangling the basis for these complex ecological and agronomical interactions.

Association mapping and the genetic basis for plant phenotypic diversity
Connecting traits to causal genes through association studies can accelarate translational research and gain insights into the underlying biological processes.
Comparative genomic and gene synteny
Comparative genomics and phylogenemocs approaches are useful for understanding complex gene duplication, deletion and rearrangement patterns. A better understanind of these events can shed a light on gene expression regulation and neofunctionalization across the genetic diversity of plants.